研究背景
近年來�,隨著電池技術(shù)蓬勃發(fā)展��,鋰離子電池(LIBs)已經(jīng)廣泛應(yīng)用于商業(yè)化便攜式電子設(shè)備����、電動汽車和可持續(xù)能源電網(wǎng)等領(lǐng)域����,其能量密度取得了顯著進(jìn)展,達(dá)到了接近300 Wh kg-1��,比傳統(tǒng)鉛酸電池的能量密度提高了5-10倍����,但仍低于行業(yè)要求的400或800 Wh kg-1。此外���,受限于鋰資源有限以及成本波動等問題����,促使研究者需要進(jìn)一步探索其他替代電池技術(shù)��。在正在研究的各種電池系統(tǒng)中�����,多價金屬電池,如鎂(Mg)�,鈣(Ca)和鋅(Zn)電池等由于其潛在的商業(yè)可行性而備受關(guān)注。Mg和Ca元素具有高豐富度�,成熟高效的冶煉技術(shù),盡管相對原子質(zhì)量高�����,但是其自身雙電子反應(yīng)使其具有優(yōu)越的理論體積容量��,確保其作為靜態(tài)儲能解決方案的有效候選材料(圖1)��,但電解質(zhì)滯后發(fā)展阻礙了這些電池的開發(fā)����,高性能電解質(zhì)不僅應(yīng)促進(jìn)負(fù)極的可逆多價金屬沉積和剝離,而且還應(yīng)支持高壓正極的可逆嵌入和脫出����,但由于多價陽離子的緩慢溶解和擴(kuò)散而產(chǎn)生的高界面阻力,使得尋找匹配的電解質(zhì)體系變得更加復(fù)雜(圖2)�?�;谏鲜鲅芯勘尘埃揪C述對Mg和Ca電池的主要失效機(jī)制進(jìn)行全面重新評估�,目的是調(diào)查以下有爭議問題:(1)理論預(yù)測的Mg/Ca金屬負(fù)極上無枝晶沉積的潛力;(2)電荷轉(zhuǎn)移和SEI擴(kuò)散的主要限制因素和(3)有機(jī)和無機(jī)SEI的聯(lián)合使用,從而提高研究者對多價金屬電池的理解�,為開發(fā)新型高性能電池體系提供重要基礎(chǔ)。
圖1. 單價和多價金屬電池的固有特性對比
文章簡介
近期���,韓國首爾大學(xué)Jang Wook Choi和高麗大學(xué)Dong-Joo Yoo等在Chemical Society Reviews上發(fā)表了題為“Insights from Li and Zn systems for advancing Mg and Ca metal batteries”的綜述文章���。本文綜述了近年來二價鎂金屬電池和鈣金屬電池的研究進(jìn)展,特別側(cè)重于與鋰金屬和鋅金屬電池進(jìn)行類比����,主要內(nèi)容涵蓋對熱力學(xué)和金屬負(fù)極形態(tài)演變的基本理解,電解質(zhì)和SEI成分之間的相關(guān)性�����,實現(xiàn)鎂和鈣可逆沉積和剝離的高效電解質(zhì)��,以及SEI特性及其與腐蝕和壽命相關(guān)性等方面�。此外,本文鼓勵研究人員通過與類似電池體系中的成功案例聯(lián)系起來�,深入研究這些新興電池領(lǐng)域,以開發(fā)高性能金屬電池體系��,對電池領(lǐng)域的長期高效發(fā)展具有重要指導(dǎo)意義。
內(nèi)容簡介
1���、鎂/鈣金屬電池的主要關(guān)注點
1.1 枝晶生長行為
理論上�,Mg由于其HCP結(jié)構(gòu)能夠抵抗枝晶形成���,并且這些枝晶不同于通常在Li中觀察到的枝晶形態(tài)(圖2和圖3)��。均勻且密集堆積的電沉積使刺穿隔膜和產(chǎn)生死金屬風(fēng)險最小�����,確保了較小比表面積�,以最大限度地減少副反應(yīng)和熱失控�。Mg較低的自擴(kuò)散能有利于平面內(nèi)沉積,與Li相比本質(zhì)上可以防止枝晶生長�����,而電流密度��、電解質(zhì)濃度和配體的存在等因素可以影響枝晶形態(tài)�����,并影響其從細(xì)顆粒到球形沉積物的演變,最終形成獨特枝晶結(jié)構(gòu)���。Ca沉積也呈現(xiàn)從理想的致密和均勻的塊狀到不太有利的枝晶和球形聚集。盡管熱力學(xué)性質(zhì)有利于在Mg和Ca金屬負(fù)極上無枝晶沉積�����,但由于各種非熱力學(xué)因素��,如低體擴(kuò)散��、鈍化層和緩慢的電荷轉(zhuǎn)移����,仍會導(dǎo)致枝晶產(chǎn)生。
圖2. 單價和多價金屬電池的理論和實驗電沉積形貌
圖3. 單價和多價金屬電池的枝晶生長預(yù)測和形貌
1.2 鈍化和微短路現(xiàn)象
與理論預(yù)測不同��,Mg和Ca金屬的枝晶形成的主要挑戰(zhàn)是陽離子絕緣鈍化層���,這與鋰金屬電池中的SEI不同����。在Mg和Ca金屬表面上�,當(dāng)電流密度低于1 mA cm-2時�����,會產(chǎn)生大于1 V的過電位��,遠(yuǎn)高于堿金屬表面上SEI鈍化層的過電位��。與Li+相比���,Mg2+的高電荷密度和Ca2+的大尺寸意味著通過離子鈍化層擴(kuò)散更具挑戰(zhàn)性,這種固有特性使得傳統(tǒng)鹽和溶劑型電解質(zhì)失效��,并促使研究者對各種鈍化消除添加劑和定制鹽進(jìn)行了廣泛研究��,但這也就降低了Mg和Ca金屬電池的成本優(yōu)勢��。近期研究希望可以確定一種單一溶劑與傳統(tǒng)亞胺基或三酸鹽配對的電解質(zhì)���,但在大多數(shù)情況下�����,這種組合仍然會導(dǎo)致高過電位和恒流循環(huán)期間電壓突降��,有時會演變成高度振蕩行為(圖4)����,可疑峰形通常被解釋為從不均勻鈍化負(fù)極中剝離初始Mg/Ca的結(jié)束,或者在循環(huán)之前從金屬-電解質(zhì)界面快速去除預(yù)吸附物質(zhì)�。然而,類比于Li和Zn金屬電池��,類似方形電壓曲線經(jīng)常被解釋為微短路�����,而Mg和Ca金屬電池中電解質(zhì)對這些電壓形狀的感應(yīng)被解釋為不可逆狀態(tài)����。在正常情況下��,這些成核過電位在整個循環(huán)過程中不斷減弱���,直到電池最終短路����。Mg和Ca金屬電池在較低電流密下表現(xiàn)出更高的過電位值��,反映了其雙電荷���、大離子半徑和高電荷密度��,在薄鈍化界面中只有少數(shù)幾個點可以進(jìn)行初始成核�����,這將電沉積限制在有限表面積內(nèi)���,并使Mg和Ca金屬電池系統(tǒng)更容易受到枝晶生長的影響���。
圖4. 金屬電池在不同循環(huán)階段的恒流充放電曲線和枝晶生長行為分析
1.3 去除天然氧化層
雖然電解質(zhì)驅(qū)動的鈍化界面在整個循環(huán)過程中控制著電沉積,但以氧化物成分為代表的自然鈍化層也顯著影響電沉積形貌����,特別是在成核階段,例如�,僅僅暴露在空氣中幾秒鐘就會顯著破壞晶體學(xué)特征。與在循環(huán)過程中形成的SEI層不同�����,天然氧化物成分長期積累��,生長成一層厚層,因此在電池制造之前去除這一層至關(guān)重要�����。氧化層問題也經(jīng)常在Mg和Ca金屬電池界討論��,因為它們具有中等到高的遷移阻力���,傳統(tǒng)LMBs的氧化層可以被Li+的高擴(kuò)散速率穿透����,而MgO呈現(xiàn)出高遷移和擴(kuò)散勢壘�,可能會阻礙具有高電荷密度的Mg2+遷移�,Mg金屬箔的硬度高,可以使用砂紙去除天然氧化層�,或者將諸如MgCl2和Cl2等吸附劑引入電池中,通過腐蝕或通過與吸附競爭在電雙層內(nèi)吸附來消除鈍化層���。相比之下���,Ca金屬無需大量表面預(yù)處理或使用苛刻電解質(zhì),在電化學(xué)過程中可能形成的天然氧化物通常不會阻礙常規(guī)的電鍍和剝離行為�����,即便需要也只需在80 mV s-1下進(jìn)行5次活化即可,Ca金屬電池的可逆性很大程度上取決于陽離子導(dǎo)電電解質(zhì)驅(qū)動的SEI層�,Ca(ClO4)2、Ca(PF6)2��、Ca(BF4)2����、Ca(TFSI)2、Ca(OTF)2等鈣鹽形成氯化鈣�、氟化物和碳酸鹽界面相的形成,這些都阻礙了陽離子的轉(zhuǎn)移��,所以僅通過調(diào)節(jié)可能無法確保Ca金屬電池的可逆性���,還需要一種形成新的間相電解質(zhì)��。
圖5. 不同金屬電沉積行為
圖6. 金屬鈍化層分析
2�����、鎂/鈣金屬電池的先進(jìn)策略
通過SEI改性策略�,可將陽離子絕緣SEI替換為陽離子導(dǎo)電SEI�����,從而使電沉積變得可行。與Li沉積不同�����,Mg/Ca沉積的擴(kuò)散動力學(xué)緩慢�,這可能導(dǎo)致電流密度超過極限電流密度。Janna等人計算了低濃度電解質(zhì)的極限電流密度值�����,并觀察到成為體擴(kuò)散限制后枝晶引起的短路現(xiàn)象�����,即在電解質(zhì)內(nèi)部Mg2+不充足的情況下�,可能會在襯底-電解質(zhì)界面迅速耗盡�����,導(dǎo)致樹枝晶生長����。Ca枝晶的生長也經(jīng)常出現(xiàn)在高充放電倍率條件,即使使用陽離子導(dǎo)電SEI,也可以觀察到獨特枝晶結(jié)構(gòu)穿過隔膜并誘導(dǎo)軟短路�����。在某些高度可逆電解質(zhì)中�����,研究者應(yīng)該充分理解和利用多價體系的電化學(xué)性能和沉積形態(tài)��,需要重點關(guān)注通過SEI層和金屬襯底附近的擴(kuò)散和電荷轉(zhuǎn)移行為�����。
圖7. Mg金屬電池電沉積研究
圖8. 不同電解質(zhì)對比研究
2.1 鋰金屬電池SEI層的基本原理
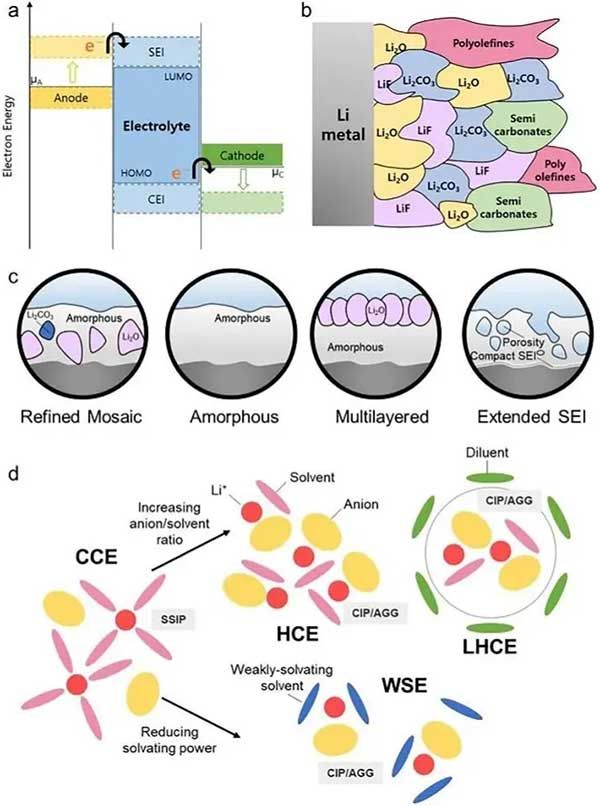
對于LMBs來說��,SEI層十分重要�����,SEI由電解質(zhì)分解形成����。盡管Mg和Ca金屬的還原電位略高于Li,但化學(xué)勢足夠高����,可以化學(xué)分解傳統(tǒng)的碳酸鹽和乙醚電解質(zhì)�����,其還原分解電位相對于在2.6至1.36 V之間��。初始循環(huán)期間的成核過電位與天然氧化層密切相關(guān)�����,長循環(huán)期間的沉積形貌是由SEI層不斷形成和擴(kuò)散所決定���。1979年,Peled引入的SEI模型是以堿金屬電池和堿土金屬電池的基礎(chǔ)�,其中SEI作為金屬負(fù)極上的保護(hù)層,由分解鹽��、溶劑分子和雜質(zhì)的積累形成����,包括有機(jī)和無機(jī)成分����,促進(jìn)離子轉(zhuǎn)移�����,同時將電極與電解質(zhì)隔離�,緩沖正在進(jìn)行的副反應(yīng)等���。SEI的形成不僅受熱力學(xué)穩(wěn)定性的影響��,還受雙電層(EDL)影響�����。SEI的形成是一個動態(tài)過程�����,與金屬電沉積競爭���,導(dǎo)致SEI不斷再生。事實上���,SEI從傳統(tǒng)的鑲嵌模型過渡到更復(fù)雜的模型����,如精細(xì)鑲嵌、無定形��、多層和擴(kuò)展的SEI模型��,這有助于推進(jìn)二價金屬電池研究�。
LIBs和LMBs系統(tǒng)需要無機(jī)SEI,包括LiF和Li2O作為成分���,以平衡高電子絕緣性和中等離子電導(dǎo)率�,這是為了減少系統(tǒng)中電解質(zhì)分解和膨脹���。電解質(zhì)的溶劑化結(jié)構(gòu)對策略通常有利于形成接觸離子對(CIP)和聚集鹽(AGG)���,這些方法為高濃度和弱溶劑化電解質(zhì)開辟了領(lǐng)域。對于Mg和Ca金屬電池來說�,與LMB系統(tǒng)相比,可以通過使用氟和氧等無機(jī)成分來實現(xiàn)電子絕緣的策略��,其陽離子的多價性質(zhì)使SEI擴(kuò)散更復(fù)雜�,Mg和Ca電池需要更強(qiáng)的離子傳輸來促進(jìn)擴(kuò)散。
圖9. 負(fù)極��、電解液和正極及產(chǎn)生SEI和CEI的相應(yīng)電子轉(zhuǎn)移過程
2.2 控制SEI層擴(kuò)散的關(guān)鍵因素
為了避免形成如CaF2的絕緣組分性成分�,Gao等人最近使用烷氧基功能化陽離子來操縱Ca2+配位環(huán)境,形成一個主要由有機(jī)基質(zhì)組成的SEI層��,類似于聚環(huán)氧乙烷�����,這種富含有機(jī)物的成分顯著促進(jìn)Ca2+擴(kuò)散�����。其他富有機(jī)原位SEI策略包括:加入硼源���,以誘導(dǎo)形成SEI中有機(jī)硼團(tuán)簇�。為了避免Mg和Ca金屬與電解質(zhì)直接接觸造成鈍化���,研究人員還制造了一層人工SEI層�����,以確保離子轉(zhuǎn)移��,在循環(huán)過程中����,這種人工SEI轉(zhuǎn)化為納米晶體,有效抑制陰離子分解�,增強(qiáng)Ca金屬的可逆電鍍和剝離。同樣�,在Mg金屬電池中,可以通過旋轉(zhuǎn)涂層���、浸漬涂層形成有機(jī)人工SEI���。雖然富無機(jī)SEI層通常被認(rèn)為不利于多價陽離子運輸,但由于它們具有陽離子絕緣性���,可采用納米級厚度的單組分或多組分無機(jī)SEI層進(jìn)行可逆沉積或剝離����。在Mg金屬電池系統(tǒng)中��,厚度為幾納米的Mg2+被巧妙地引入到Mg負(fù)極表面��,以形成Mg2+導(dǎo)電層。在Ca金屬體系中�����,薄CaO主導(dǎo)層可以實現(xiàn)離子轉(zhuǎn)移�����,并且通過控制非均質(zhì)性可以改變電導(dǎo)率�����。大多數(shù)無機(jī)策略取決于確保形成一個薄SEI層���,以獲得高可逆性和低過電位,有時涉及硼或氧作為共同成分����。采用硼所產(chǎn)生的優(yōu)越性能的確切機(jī)制需要進(jìn)一步研究,但可以基于對LMB系統(tǒng)的理解�����,硼的膨脹性質(zhì)加上電子數(shù)量的減少可能會通過與無機(jī)部分形成稀疏的聚合物網(wǎng)絡(luò)來緩解具有高電荷密度的多價陽離子滲透�����,這可能有助于陽離子轉(zhuǎn)移,硼不僅有利于形成有機(jī)硼團(tuán)簇�,而且有助于最終形成薄SEI層。
圖10. Ca和Mg金屬電池SEI組分調(diào)控
圖11. 硼元素引入金屬電池
2.3 優(yōu)化電荷傳遞動力學(xué)
為最小化SEI阻力�����,傳統(tǒng)方法主要使用大尺寸陰離子���,如鹵素和烷氧硼酸鹽結(jié)構(gòu)�,由于這些陰離子與金屬陽離子的配位較弱��,有利于增強(qiáng)脫溶作用����。然而,傳統(tǒng)鹵素和硼基鹽由于其低負(fù)極穩(wěn)定性而與高壓正極不相容��,且以烷氧硼酸鹽為代表的高相容電解質(zhì)通常合成復(fù)雜�����。因此�����,目前正在進(jìn)行大量的研究,以探索使用結(jié)構(gòu)穩(wěn)定和容易獲得的常規(guī)鹽��,特別是亞胺類和三氟酸鹽類�����。SEI改性通常針對界面來避免降低體電阻���,而控制電荷轉(zhuǎn)移通常需要改變電解質(zhì)的溶劑化結(jié)構(gòu),從而進(jìn)一步影響性能�。例如,Mg和Ca金屬電池通常需要具有強(qiáng)結(jié)合能力的高供體溶劑�����,以溶解傳統(tǒng)相容鹽的緊密結(jié)合的陽離子-陰離子復(fù)合物����,否則,電解質(zhì)濃度將被限制在0.5 M以下���,導(dǎo)致低離子電導(dǎo)率��。為了解決這個問題����,最近的研究引入了一些方法來平衡鹽和溶劑之間的配位,使陽離子能夠很容易地從陰離子中分離出來��。與引入或合成新的溶劑/鹽相比�,更保守的方法是將常規(guī)鹽和溶劑的簡單混合物作為Mg或Ca電解質(zhì)的助溶劑或助鹽,并削弱陽離子第一溶劑化層的配位�����。Zn金屬電池也采用了類似策略����,將溶劑化的H2O部分替換為引入比H2O給體數(shù)更高的有機(jī)分子,如二甲亞砜(DMSO)(DMSO)等���。與LMBs中使用的非極性聚乙烯和聚丙烯相比����,在Mg和Zn金屬電池系統(tǒng)中����,對金屬具有親和力���,并且易于容納金屬沉積物。此外����,必須認(rèn)識到電池系統(tǒng)中電極實際與隔膜接觸的環(huán)境與電極間距更遠(yuǎn)的替代配置環(huán)境之間的區(qū)別,還需要關(guān)注最小化界面阻力和改進(jìn)隔膜策略的研究�����,以準(zhǔn)確確定真實沉積行為�。
圖12. 優(yōu)化電荷傳輸策略
3��、Ca和Mg金屬電池的聯(lián)系
在LMBs中�,庫侖效率高于90%的降解通常分為兩種途徑:SEI生長和死鋰的形成。值得注意的是����,除了LMBs的庫侖效率為95%之外,特定相間組分的豐度或稀缺性通常會直接影響性能���。Mg和Ca金屬電池兼容的電解質(zhì)還未完全深入研究以確定其長期循環(huán)屬性和沉積形態(tài)�����,所以表面和間相化學(xué)的原位表征將是未來研究重點�����。先進(jìn)表征技術(shù)包括:
(1)X射線光電子能譜�����、低溫透射電鏡����、核磁共振等技術(shù)能夠在局部區(qū)域區(qū)分作為SEI組分的Li+和表面的金屬Li0。
(2)原位環(huán)境透射電子顯微鏡���、光學(xué)顯微鏡和X射線顯微斷層掃描主要提供了對SEI形態(tài)和厚度的直接觀測��。
(3)滴定氣相色譜法可以準(zhǔn)確量化未反應(yīng)金屬Li0對非活性鋰含量的貢獻(xiàn)與電解質(zhì)分解量的比例�����。這些技術(shù)可以加深對SEI層的組成和結(jié)構(gòu)的更深入的理解����,豐富對電池退化的認(rèn)識����,將能夠促進(jìn)Mg和Ca有效集成到下一代儲能系統(tǒng)中����。
圖13. 單價和多價金屬電池關(guān)于SEI和電解質(zhì)的最新理解
3.1 SEI膨脹
最近��,Zhang等人采用了一種改進(jìn)的薄膜玻璃化技術(shù)來保持液體電解質(zhì)完整性�,使其能夠提高顯微鏡和光譜分析電池樣品的真實性(圖14),觀測發(fā)現(xiàn)SEI實際上進(jìn)入了一個膨脹狀態(tài)����,這與普遍假設(shè)相反。無論所選擇的電解質(zhì)如何���,所觀察到的膨脹現(xiàn)象的普遍性對SEI內(nèi)的離子傳輸和后續(xù)溶劑分解具有重要意義����。溶脹模型挑戰(zhàn)了前一模型的有效性�����,溶脹現(xiàn)象實際上影響了溶劑支撐下Li+擴(kuò)散���,傳統(tǒng)模型通常是基于源自LIBs體系的傳統(tǒng)假設(shè)�����,即陽離子通過固體擴(kuò)散通過無機(jī)組分傳輸���,這無法準(zhǔn)確代表膨脹SEI行為。類似誤解也出現(xiàn)在Mg和Ca金屬電池系統(tǒng)中����,這說明確定膨脹存在的必要性,考慮到溶劑衍生的SEI組分的鈍化性質(zhì)��,預(yù)成型SEI層中的膨脹程度可能會破壞Mg和Ca金屬的可逆性����,從而導(dǎo)致過電位的持續(xù)增加。因此���,在設(shè)計良好SEI層配制電解質(zhì)時����,應(yīng)考慮膨脹效應(yīng)��,以延長SEI可逆性��。
圖14. 冷凍電鏡觀測SEI膨脹現(xiàn)象
3.2 SEI溶解
SEI中陰離子衍生的無機(jī)物質(zhì),在電解質(zhì)中溶解度低���,在形成后會發(fā)生顯著溶解��。因此�,陰離子衍生成分(如LiF)可用于評估SEI在不同電解質(zhì)中的溶解度��。然而�����,隨著分析技術(shù)的發(fā)展����,需要進(jìn)一步思考。Huang等人觀察到����,由于LiF在電解質(zhì)中溶解度適中�����,傾向于在電極表面形成大顆粒�,而不是傳統(tǒng)上公認(rèn)的致密SEI層��。這一觀點與Shadike等人的發(fā)現(xiàn)相反�,他們在另一種電解質(zhì)中直接觀察到SEI中不溶的LiF納米晶����,這意味著SEI的溶解度取決于相間組分和電解質(zhì)類型。這些研究表明���,由于SEI的溶脹和溶解的綜合影響�����,僅根據(jù)其組成來評估SEI的功能對于高性能電解質(zhì)不太可行��,考慮到大多數(shù)Mg����、Ca金屬電池的電解液濃度遠(yuǎn)低于1 M�,有利于控制溶解程度,確保了不必要的成分被溶解����,而有益的成分保留下來,最終形成薄而緊湊的SEI。所以��,對Mg和Ca金屬電池的研究可能側(cè)重于溶解有機(jī)成分���,以形成更緊湊但中等離子導(dǎo)電性的無機(jī)SEI���,或?qū)⒋髩K不可滲透的無機(jī)物質(zhì)溶解成納米晶體粒子可能是另一個可行選擇。
3.3 腐蝕現(xiàn)象
電解液和鋰的持續(xù)消耗也對邁向商業(yè)化水平的道路構(gòu)成了重大障礙����。Boyle等人最近研究表明,具有高庫侖效率的電解質(zhì)有時具有較差的腐蝕性能���。根據(jù)這一觀點�,觀察到以前已知的示例性電解質(zhì)的大DCE主要歸因于初始SEI的溶脹和溶解����,分別導(dǎo)致了緊湊型SEI和擴(kuò)展型SEI的生長。Kwon等人指出���,僅存在無機(jī)物并不足以抑制電解質(zhì)通過SEI擴(kuò)散��,有必要進(jìn)行結(jié)構(gòu)調(diào)整�����,以減少SEI中的顆粒間空間����,從而最終提高抑制Li腐蝕的鈍化性�����。在實現(xiàn)致密金屬沉積后�,Mg/Ca金屬電池具有高庫侖效率,最小化腐蝕將是優(yōu)化長期存儲的下一個目標(biāo)�����。特別是在Mg金屬電池中����,微量水引起的腐蝕效應(yīng)經(jīng)常超過電化學(xué)鈍化的影響。因此����,未來的研究不僅應(yīng)關(guān)注可逆性,還應(yīng)包括對腐蝕影響的分析�����。
3.4 貧電解質(zhì)
眾所周知,提高重量和體積能量密度將成為多價金屬電池研究領(lǐng)域的目標(biāo)�,貧電解質(zhì)設(shè)計是這一目標(biāo)的核心。多價金屬電池和鋰離子電池之間的電解質(zhì)容量比的巨大差異表明����,在循環(huán)期間,需要大量的電解質(zhì)來補(bǔ)償其不可逆轉(zhuǎn)的損失��。在LMBs中����,電解質(zhì)消耗導(dǎo)致的性能下降已被確定為軟包電池的關(guān)鍵問題。因此�����,在基于其他離子的系統(tǒng)中����,有必要預(yù)先考慮并解決類似問題,特別是在腐蝕性方面����,探索抗嚴(yán)重腐蝕的螯合劑將有助于其商業(yè)化進(jìn)程�。
全文總結(jié)
綜上所述�����,多價金屬電池的發(fā)展���,特別是使用Mg和Ca金屬負(fù)極的電池,既帶來了巨大機(jī)遇�,也帶來了重大挑戰(zhàn)。盡管由于Mg和Ca的天然豐度��,以及其在理論上具有很高的體積能量密度和成本效益�����,但它們的實際應(yīng)用方面受到幾個關(guān)鍵因素阻礙�,但可以通過Li和Zn金屬電池系統(tǒng)的認(rèn)識來幫助克服這些障礙:
(1)枝晶生長與沉積形態(tài);
(2)原生氧化層和鈍化;
(3)電解質(zhì)相容性與SEI形成;
(4)電荷轉(zhuǎn)移和解離過程;
(5)SEI的膨脹和溶解;
(6)腐蝕和老化;
(7)貧電解質(zhì)設(shè)計。
總的來說����,Mg和Ca金屬電池的發(fā)展取決于多方面因素,包括沉積形態(tài)���、SEI形成���、電荷轉(zhuǎn)移動力學(xué)和耐腐蝕性�����,通過利用更成熟的Li和Zn電池的相關(guān)見解��,結(jié)合電解質(zhì)化學(xué)和界面工程的持續(xù)創(chuàng)新����,多價金屬電池的商業(yè)化發(fā)展可以逐步推進(jìn)����,為下一代儲能系統(tǒng)提供有前途的解決方案(表1)。
表1 各種金屬負(fù)極電池中設(shè)計電解質(zhì)和SEI層的要點
文獻(xiàn)信息
Insights from Li and Zn systems for advancing Mg and Ca metal batteries. Chem. Soc. Rev., 2024. (DOI: 10.1039/d4cs00557k);https://doi.org:/10.1039/d4cs00557k
免責(zé)聲明
本文內(nèi)容來源于“能源學(xué)人”�,版權(quán)歸原作者所有,發(fā)文目的在于傳遞更多信息����,并不代表本公眾號贊同其觀點和對其真實性負(fù)責(zé)。如涉及作品內(nèi)容�、版權(quán)和其它問題,請來電或致函告之�,我們將及時給予處理!
 606
606
 0
0 

測和形貌")
階段的恒流充放電曲線和枝晶生長行為分析")



對比研究")
控")

化電荷傳輸策略")
于SEI和電解質(zhì)的最新理解")
象")
極電池中設(shè)計電解質(zhì)和SEI層的要點")
 分享 0
分享 0
 舉報 0
舉報 0
 收藏 0
收藏 0
 反對 0
反對 0
 點贊 0
點贊 0

 中冶有色技術(shù)平臺
中冶有色技術(shù)平臺






































































