全部
▼
搜索
熱搜:
位置:中冶有色 >
> 高效除氟材料及其制備方法
 271
編輯:中冶有色技術(shù)網(wǎng)
來(lái)源:中科格潤(rùn)(唐山)環(huán)境技術(shù)有限公司
271
編輯:中冶有色技術(shù)網(wǎng)
來(lái)源:中科格潤(rùn)(唐山)環(huán)境技術(shù)有限公司
[0058]表1含氟廢水中殘余氟離子濃度結(jié)果

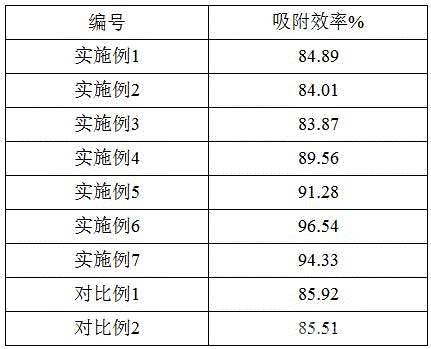
[0061]表2 10次循環(huán)后吸附效率結(jié)果

 分享 0
分享 0
 舉報(bào) 0
舉報(bào) 0
 收藏 0
收藏 0
 反對(duì) 0
反對(duì) 0
 點(diǎn)贊 0
點(diǎn)贊 0

 中冶有色技術(shù)平臺(tái)
中冶有色技術(shù)平臺(tái)微細(xì)粒礦物選礦技術(shù)大會(huì)")
 2025年03月25日 ~ 27日
2025年03月25日 ~ 27日 新大會(huì)") 2025年03月28日 ~ 30日
2025年03月28日 ~ 30日 創(chuàng)新大會(huì)") 2025年03月28日 ~ 30日
2025年03月28日 ~ 30日 核材料產(chǎn)學(xué)合作高峰論壇") 2025年04月11日 ~ 13日
2025年04月11日 ~ 13日 ") 2025年04月24日 ~ 27日
2025年04月24日 ~ 27日 
 京公網(wǎng)安備 11010702002294號(hào)
京公網(wǎng)安備 11010702002294號(hào)










































































