全部
▼
搜索
熱搜:
位置:中冶有色 >
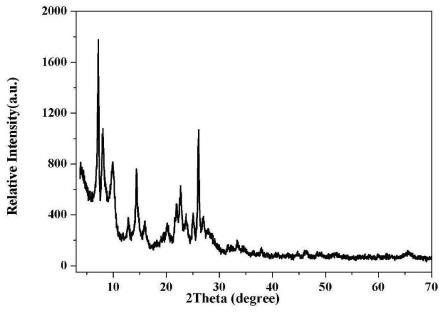
> 分子篩及其制備方法和應(yīng)用與流程
 587
編輯:中冶有色技術(shù)網(wǎng)
來源:中國石油化工股份有限公司上海石油化工研究院
587
編輯:中冶有色技術(shù)網(wǎng)
來源:中國石油化工股份有限公司上海石油化工研究院
 分享 0
分享 0
 舉報 0
舉報 0
 收藏 0
收藏 0
 反對 0
反對 0
 點贊 0
點贊 0



 2025年03月20日 ~ 22日
2025年03月20日 ~ 22日 新大會") 2025年03月21日 ~ 23日
2025年03月21日 ~ 23日 術(shù)會議") 2025年03月28日 ~ 30日
2025年03月28日 ~ 30日 用技術(shù)交流會") 2025年03月29日 ~ 31日
2025年03月29日 ~ 31日 用技術(shù)交流會") 2025年04月11日 ~ 13日
2025年04月11日 ~ 13日 












































































