全部
▼
搜索
熱搜:
 872
編輯:中冶有色技術網
來源:侯志全,郭萌,劉雨溪,鄧積光,戴洪興
872
編輯:中冶有色技術網
來源:侯志全,郭萌,劉雨溪,鄧積光,戴洪興
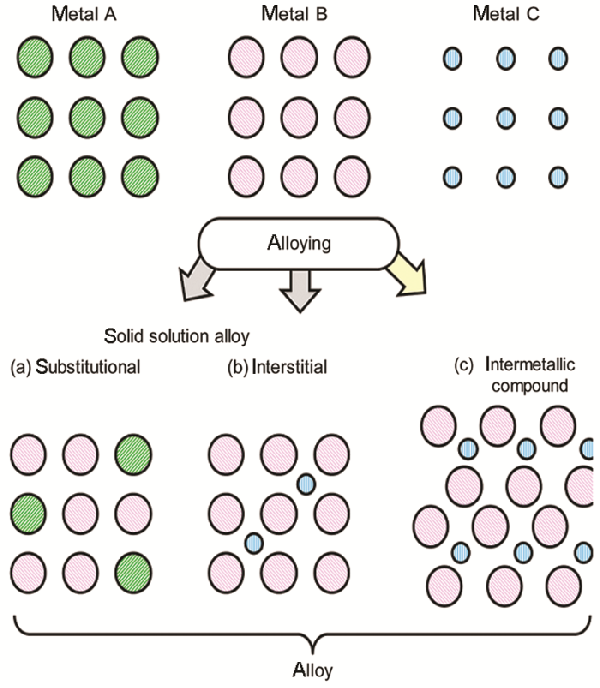



| Intermetallic compound | Synthesis method | Crystal phase | Particle size | Reaction condition | Catalytic performance | Ref. |
|---|---|---|---|---|---|---|
| Pd5Ga3 | Chemical reduction | Orthorhomibc | 5.3 nm | 0.5% CH4, 4% O2, N2 balance; space velocity (SV): 80000 mL/(g·h) | T90% is lower to 372oC, the special reaction rate of Pd is 23.32×10-6 mol/(gPd s) at 290oC. | [12] |
| Ni3Ga, Ni3Sn2 | Chemical reduction | Cubic | 3.5~7.5 nm | Pretreated with H2; 1 mmol substrate and 0.5 mmol n-dodecane; H2: 500 kPa; 1300 r/min | After 13 h continuous reaction, the conversion of various types of alkyne reached more than 90%, and the selectivity of olefins was over 94%. | [15] |
| PdmMn (M=Ge, In, Sn, Zn) | Gas-phase reduction | - | - | 12.5% butylene, butylene: O2=1:1, He (balance); total gas flow rate: 120 mL/min | PdIn, PdBi or Pd3Fe catalysts show high selectivity for 1,3-Butadiene and 1-butene (more than 50%) and high yield. | [20] |
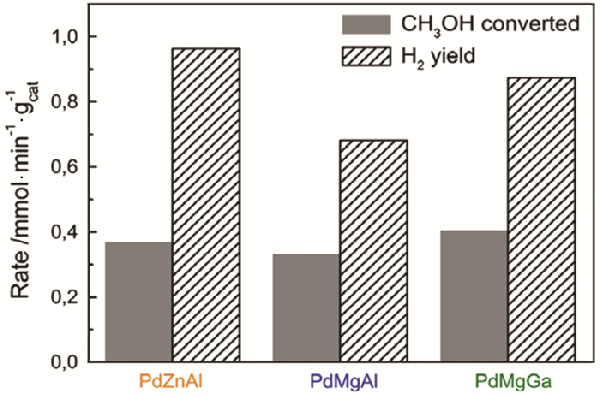
| Pd2Ga and PdZn | Gas-phase reduction | Cubic | - | H2O/CH3OH=1.0, total gas flow rate: 26 mL/min, methanol concentration: 28.4% | PdZnAl exhibited the best catalytic activity, with 87% hydrogen selectivity at 250oC and hydrogen generation rate of 964 μmol/(g·min). | [21] |
| Ni3Sn, Ni3Sn2, Ni3Sn2, Ni3Sn4 | Arc melting | - | 25~38 μm | The partial pressures of acetylene and hydrogen are 2.7 and 13 kPa, respectively. | The acetylene conversion rates of Ni3Sn, Ni3Sn2 and Ni3Sn4 are 2.3×10-6 mol/(g·s), 0.6×10-6 mol/(g·s) and less than 0.001×10-6 mol/(g·s), respectively. | [22] |
| Pt3Sn | Polyol process | - | 5.2±1.0 nm | O2/CO=6:1; room temperature | The initiation temperature of CO oxidation on Pt3Sn is lower than that on the Pt catalyst. | [31] |
| Pt3Ti | Chemical reduction | Cubic | 2.5 nm | 2 % CO, 1% O2, 97% He, space velocity (SV): 120000 mL/(g·h) | The ignition temperature of CO oxidation on Pt3Ti catalyst is 125oC, which is lower than that on single Pt catalyst. | [43] |
| Ni2Si, NiSi or NiSi2 | Chemical vapor deposition | Cubic | 3~4 nm | H2/CO=3:1, Ar balance, space velocity (SV): 48000 mL/(g·h) | The activity of CO methanation on Ni-Si catalyst is much higher than that on single nickel catalyst, with enhanced stability of nickel sintering resistance at high temperature (500~600oC). | [49] |
| NiZn | Thermal annealing | Cubic | 20~32 μm | methanol:H2O=1:1; 0.01 mL/min, N2: 13.2 mL/min, He: 1.6 mL/min | NiZn catalyst has good catalytic performance for methanol reforming (80% conversion at 550oC) and good hydrogen selectivity (70%). | [57] |


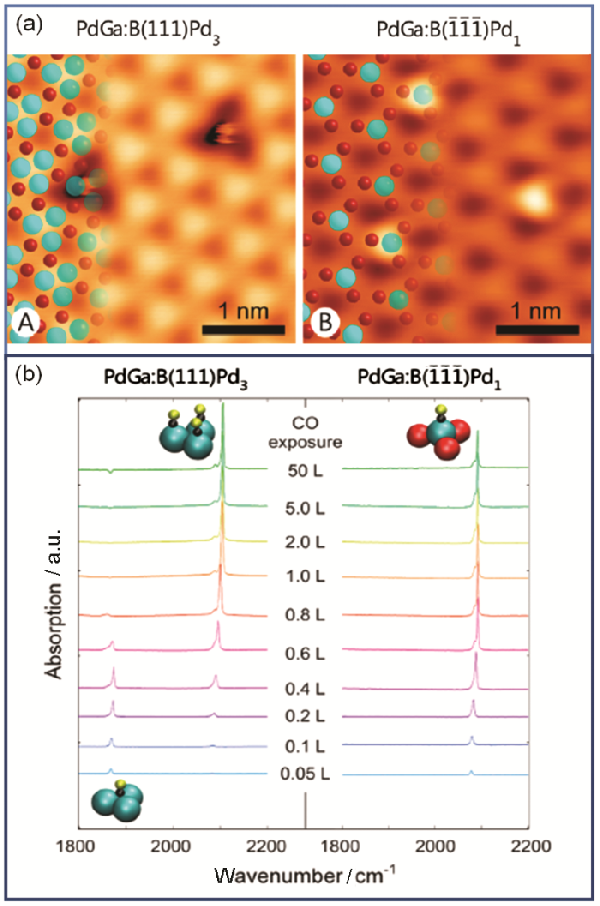

 分享 0
分享 0
 舉報 0
舉報 0
 收藏 0
收藏 0
 反對 0
反對 0
 點贊 0
點贊 0









































































 中冶有色技術平臺
中冶有色技術平臺
 2025年03月28日 ~ 30日
2025年03月28日 ~ 30日 


新發(fā)展論壇")




